
Durante la pandemia di Covid, il governo degli Stati Uniti ha speso miliardi di dollari in quasi 400 prodotti destinati a proteggere, diagnosticare e curare centinaia di milioni di persone, tutti con l’etichetta “EUA” o “Autorizzazione all’uso di emergenza”.
Ma cosa significa effettivamente EUA?
Anche prima di rispondere a questa domanda, e per comprendere la posizione dell'EUA rispetto ad altri percorsi di autorizzazione o approvazione di prodotti medici, è utile esaminare cosa non è l'EUA:
L'EUA non è una designazione per un prodotto sperimentale sottoposto a sperimentazione clinica
Se capiamo solo una cosa sull’EUA dovrebbe essere questa: l’EUA non si applica a un prodotto sottoposto a una sperimentazione clinica disciplinata dalle normative FDA o da altri requisiti legali.
Inoltre, l’EUA non è la stessa cosa dell’Expanded Access Use (EAU), spesso chiamato accesso “uso compassionevole”, che si applica alla concessione ai pazienti affetti da malattie gravi e incurabili l’accesso a prodotti sperimentali prima che siano completamente approvati.
Questa tabella da un Presentazione FDA-CDC 2020 riassume le differenze tra i prodotti sottoposti a studi clinici, i prodotti forniti ai pazienti attraverso un accesso “compassionevole” ampliato e i prodotti autorizzati tramite EUA:
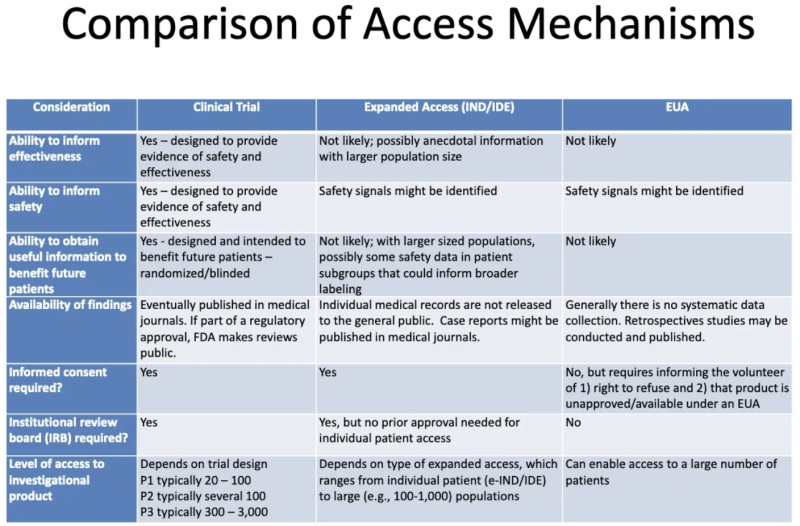
Ecco cosa ci dice questa tabella sull'EUA:
- È improbabile che il processo di concessione dell'EUA generi informazioni sull'efficacia di un prodotto.
- Il processo di concessione dell’EUA non è progettato per fornire prove di sicurezza o efficacia, ma potrebbero essere identificati segnali di sicurezza.
- È improbabile, una volta che un prodotto viene concesso con l'EUA e somministrato ad alcuni pazienti, che si ottengano informazioni utili a beneficio di eventuali futuri pazienti.
- Non esiste una raccolta sistematica di dati sull'efficacia o sulla sicurezza dell'EUA e nessun dato viene pubblicato su riviste mediche come parte del processo di approvazione normativa.
- Non è richiesto il consenso informato, ma i pazienti che si offrono volontari per assumere il prodotto devono essere informati che possono rifiutarsi e che il prodotto non è approvato/disponibile ai sensi dell'EUA.
- Non è richiesto alcun comitato di revisione istituzionale (IRB). [IRB è un comitato che dovrebbe proteggere il benessere dei soggetti umani negli studi clinici]
Per chiarire ancora di più quanto l'EUA sia separata da qualsiasi normale processo di approvazione, in a 2009 Pubblicazione dell'Istituto di Medicina delle Accademie Nazionali, troviamo questa affermazione:
È importante riconoscere che una EUA non fa parte del percorso di sviluppo; è un'entità completamente separata che viene utilizzata solo durante le situazioni di emergenza e non fa parte del processo di approvazione del farmaco. (pag. 28)
Per riassumere:
È improbabile che il processo di concessione dell'EUA di un prodotto generi prove di sicurezza o efficacia. Una volta che un prodotto ottiene l'EUA e viene somministrato ai pazienti, è improbabile che si ottengano informazioni utili a beneficio dei pazienti futuri, poiché non esiste una raccolta sistematica di dati sull'efficacia o sulla sicurezza.
Sulla base di tutte queste informazioni molto chiare provenienti dal CDC/FDA e dall’IMNA, sarebbe giusto concludere che l’autorizzazione all’uso di emergenza è un processo che dovrebbe essere applicato con molto giudizio e solo in casi di gravi emergenze.
Ora diamo un'occhiata a quali tipi di situazioni di emergenza l'EUA è legalmente progettata per affrontare.
L'EAU è pensato per le emergenze WMD
Le leggi che consentono il “Meccanismo di accesso” dell’EUA sopra descritto sono state redatte per casi di emergenze estreme e immediate che coinvolgono armi di distruzione di massa (WMD), denominate anche agenti CBRN (chimici, biologici, radiologici, nucleari).
Ecco come funziona la Food & Drug Administration (FDA) descrive i suoi poteri EUA:
Sezione 564 della legge FD&C (21 USC 360bbb-3) consente alla FDA di rafforzare la tutela della salute pubblica contro gli agenti biologici, chimici, nucleari e radiologici.
Con questa autorità EUA, la FDA può contribuire a garantire che le contromisure mediche possano essere utilizzate in caso di emergenza per diagnosticare, trattare o prevenire malattie o condizioni gravi o potenzialmente letali causate da agenti biologici, chimici, nucleari o radiologici quando non esistono adeguate misure approvate. e le alternative disponibili (tra gli altri criteri).
Questi poteri dell’EUA sono stati concessi nel 2004 in circostanze molto specifiche legate alla preparazione agli attacchi da parte di agenti CBRN.
Come spiegato nella fattura sanitaria della legge di Harvard,
Alla fine, è stata la guerra al terrorismo a dare origine all’autorizzazione all’uso di emergenza. Dopo gli eventi dell'11 settembre 2001 e i successivi attacchi postali all'antrace, il Congresso ha promulgato la Legge sul progetto Bioshield del 2004.
I record indica che il Congresso si è concentrato specificamente sulla minaccia del bioterrorismo, non sulla preparazione per una pandemia naturale.
Dato un tipo così ristretto di situazione di emergenza veramente estrema che coinvolge un attacco di armi di distruzione di massa, è comprensibile il motivo per cui il “meccanismo di accesso” dell’EUA non richiede molta supervisione normativa o aderenza a standard di produzione o di sperimentazione clinica.
Quindi cosa richiede effettivamente il meccanismo di accesso all’EUA?
I 3 passaggi per l'autorizzazione all'uso di emergenza (EUA)
Perché venga concessa l'EUA a un prodotto medico devono verificarsi tre cose:
- Il Segretario per la Sicurezza Nazionale, il Segretario alla Difesa o il Segretario per la Salute e i Servizi Umani deve stabilire se esiste un’emergenza che comporta un attacco o una minaccia di attacco con un agente CBRN o una malattia causata da tale agente.
- La FDA deve assicurarsi di soddisfare quattro “criteri statutari” quando emette l’EUA.
- La FDA deve “imporre determinate condizioni richieste” nell’EUA.
Fase 1 dell'EUA: dichiarazione di un'emergenza CBRN
La dichiarazione di emergenza per l'EUA è separata e non correlata a qualsiasi altra dichiarazione di emergenza che potrebbe essere rilasciata dal Presidente, dal Segretario dell'HHS o da chiunque altro. Deve essere emesso specificatamente allo scopo di attivare l'EUA e può essere revocato o prorogato indipendentemente da qualsiasi altra dichiarazione di emergenza.
Ecco cosa afferma la legge dell'EUA sono i quattro possibili scenari per l’attivazione del “meccanismo di accesso” all’EUA:
- una determinazione da parte del Segretario per la Sicurezza Nazionale che esiste un'emergenza interna, o un potenziale significativo per un'emergenza interna, che comporta un rischio elevato di attacco con uno o più agenti biologici, chimici, radiologici o nucleari;
- una decisione da parte del Segretario della Difesa che esiste un'emergenza militare, o un potenziale significativo per un'emergenza militare, che comporta un rischio maggiore per Unito stati forze militari, compreso il personale che opera sotto l'autorità del Titolo 10 o del Titolo 50, di attacco con:
- uno o più agenti biologici, chimici, radiologici o nucleari; O
- uno o più agenti che potrebbero causare, o essere altrimenti associati a, un rischio specifico e di imminente pericolo di vita per United stati forze militari;
- una determinazione da parte del segretario [della sanità e dei servizi umani] che esiste un'emergenza sanitaria pubblica, o un potenziale significativo per un'emergenza sanitaria pubblica, che colpisce, o ha un potenziale significativo di influenzare, la sicurezza nazionale o la salute e la sicurezza di Unito stati cittadini che vivono all'estero e che coinvolgono uno o più agenti biologici, chimici, radiologici o nucleari, o una malattia o condizione che può essere attribuibile a tale agente o agenti; O
- l'identificazione di una minaccia materiale ai sensi della sezione 319F–2 del Legge sul servizio sanitario pubblico [42 USC 247d-6b] sufficienti a pregiudicare la sicurezza nazionale o la salute e la sicurezza Unito stati cittadini residenti all'estero.
EUA Fase 2. Soddisfare i criteri statutari
Una volta che uno dei segretari ha dichiarato che esiste un’emergenza che giustifica l’EUA, ci sono altri quattro “criteri statutari” che devono essere soddisfatti affinché la FDA possa rilasciare l’EUA. Ecco come la FDA spiega questi requisiti:
- Malattia o condizione grave o pericolosa per la vita
Affinché la FDA possa rilasciare un EUA, gli agenti CBRN a cui si fa riferimento nella dichiarazione EUA del segretario dell'HHS devono essere in grado di causare una malattia o una condizione grave o pericolosa per la vita.
- Prove di efficacia
I prodotti medici che possono essere presi in considerazione per un EUA sono quelli che "potrebbero essere efficaci" per prevenire, diagnosticare o trattare malattie o condizioni gravi o pericolose per la vita che possono essere causate da uno o più agenti CBRN identificati nella dichiarazione di conformità del segretario dell'HHS. emergenza o minaccia di emergenza ai sensi della sezione 564(b).
Lo standard “può essere efficace” per gli EUA prevede un livello di prova inferiore rispetto allo standard di “efficacia” utilizzato dalla FDA per l’approvazione dei prodotti. La FDA intende valutare caso per caso la potenziale efficacia di un possibile prodotto EUA utilizzando un'analisi rischio-beneficio, come spiegato di seguito.
[GRESETTO AGGIUNTO]
- Analisi rischio-beneficio
Un prodotto può essere preso in considerazione per un EUA se il Commissario determina che i benefici noti e potenziali del prodotto, quando utilizzato per diagnosticare, prevenire o trattare la malattia o condizione identificata, superano i rischi noti e potenziali del prodotto.
Nel determinare se i benefici noti e potenziali del prodotto superano i rischi noti e potenziali, la FDA intende guardare alla totalità delle prove scientifiche per effettuare una determinazione complessiva del rapporto rischio-beneficio. Tali prove, che potrebbe sorgere da una varietà di fonti, Può includere (ma non limitato a): risultati di studi clinici nazionali ed esteri, dati di efficacia in vivo da modelli animali e dati in vitro, disponibile per l'esame della FDA. La FDA valuterà anche la qualità e la quantità del prove disponibili, considerato lo stato attuale delle conoscenze scientifiche.
[GRESETTO AGGIUNTO]
- Nessuna alternativa
Affinché la FDA possa rilasciare un EUA, non deve esistere un'alternativa adeguata, approvata e disponibile al prodotto candidato per diagnosticare, prevenire o trattare la malattia o la condizione. Un potenziale prodotto alternativo può essere considerato “non disponibile” se le scorte dell’alternativa approvata non sono sufficienti per soddisfare pienamente il bisogno di emergenza.
EUA Fase 3. Imposizione delle condizioni richieste
Una volta ottenuta la dichiarazione di emergenza specifica dell’EUA e una volta che la FDA stabilisce che il prodotto può essere efficace e che qualunque prova disponibile dimostri che i suoi benefici superano i rischi, esiste un ulteriore livello di regolamentazione correlata.
Ecco come a Rapporto 2018 del Servizio di ricerca del Congresso sull'EUA spiega questo:
L'FFDCA §564 impone alla FDA di imporre determinate condizioni richieste in un EUA e consente ulteriori condizioni discrezionali ove appropriato. Le condizioni richieste variano a seconda che l'EUA riguardi un prodotto non approvato o un uso non approvato di un prodotto approvato. Per un prodotto non approvato, le condizioni d'uso devono:
(1) garantire che gli operatori sanitari che somministrano il prodotto ricevano le informazioni richieste;
(2) garantire che le persone a cui viene somministrato il prodotto ricevano le informazioni richieste;
(3) prevedere il monitoraggio e la segnalazione degli eventi avversi associati al prodotto; E
(4) prevedere la tenuta dei registri e la rendicontazione da parte del produttore.
Conclusione
Come osservato in questo articolo, la FDA/CDC riconosce chiaramente che è improbabile che il processo di concessione dell'autorizzazione all'uso di emergenza (EUA) generi informazioni sull'efficacia o sulla sicurezza di un prodotto. Quando guardiamo la lettera della legge che disciplina l’EUA, vediamo che questa è, effettivamente, una valutazione corretta.
La legge EUA non impone standard legali o normativi che potrebbero determinare se un prodotto è sicuro o efficace. Gli unici standard sono se la FDA ritiene che il prodotto possa essere efficace e che i suoi benefici noti superino i suoi danni noti. Se non sono noti danni o benefici noti, poiché il prodotto non è mai stato sottoposto al processo di approvazione del farmaco, la FDA può utilizzare qualsiasi informazione o standard scelga per prendere tale decisione.
Da tutto ciò consegue che un'azienda il cui prodotto è candidato all'EUA può tentare di dimostrare la sicurezza e/o l'efficacia del prodotto con qualunque mezzo scelga. L’esistenza di un simile tentativo (che si tratti di una sperimentazione clinica o di un altro meccanismo di raccolta dati) e il modo in cui tale tentativo viene condotto dipendono dall’azienda. Nulla nella legge EUA si applica al modo in cui l'azienda progetta, conduce o analizza studi o altri meccanismi di raccolta dati che sceglie di perseguire.
Applicato ai prodotti Covid questo significa:
- Non sono stati richiesti dati sulla sicurezza o sull’efficacia provenienti da studi clinici affinché i prodotti Covid potessero ricevere l’EUA.
- Tutti gli studi clinici a cui si fa riferimento nel processo EUA sono stati condotti senza standard normativi legalmente applicabili.
- Quando scopriamo che questi prodotti mancano di efficacia o sicurezza, non è una sorpresa. È un risultato altamente probabile del processo.
- Non sono disponibili dati provenienti dal processo EUA su cui basare decisioni non EUA sulla sicurezza o sull'efficacia del prodotto. Pertanto, qualsiasi utilizzo del prodotto al di fuori dell’EUA richiederebbe di sottoporsi fin dall’inizio al processo di approvazione legale per i normali prodotti medici.
Maggiori informazioni sul processo di approvazione dei vaccini anti-Covid qui.
Ripubblicato dall'autore substack
Pubblicato sotto a Licenza internazionale Creative Commons Attribution 4.0
Per le ristampe, reimpostare il collegamento canonico all'originale Istituto di arenaria Articolo e Autore.








