

Negli ultimi decenni della mia carriera, ho trascorso innumerevoli ore a lavorare per proteggere gli americani facendo ricerche sulla sicurezza dei farmaci. La mia istruzione e carriera mi hanno portato a frequentare una mezza dozzina di università, Big Pharma e la FDA sotto tre amministrazioni presidenziali. La sicurezza dei farmaci considera il motivo per cui un individuo può assumere un prodotto farmaceutico e non avere eventi avversi, mentre un altro individuo potrebbe assumere lo stesso prodotto ma avere reazioni avverse fino alla disabilità permanente o alla morte. Per impostazione predefinita, lo studio della sicurezza dei farmaci considera anche gli aspetti non clinici della produzione e della qualità dei farmaci.
Poiché la qualità dei farmaci è un fattore essenziale nella valutazione della sicurezza dei farmaci, il mio percorso per proteggere gli americani mi ha portato a concettualizzare e fondare il primo “farmacia analitica" ha la missione di verificare scientificamente i prodotti farmaceutici provenienti da luoghi come l'India e la Cina prima di distribuirli ai pazienti. Sfortunatamente, il perseguimento della generosità rispetto all’etica e alla protezione dei pazienti ha portato a un impegno da parte della gestione finanziaria dell’azienda estensivo Violazioni della FDA ed essere accusato dai giudici di fare false affermazioni scientifiche (tutto ciò è avvenuto casualmente dopo la mia uscita).
Senza una conferma esterna della qualità del farmaco, gli americani dipendono completamente dalla FDA e dai produttori per valutare e confermare la purezza del prodotto. È stato dimostrato che la sicurezza dei farmaci è un problema degno di nota quando si tratta di iniezioni di mRNA Covid. Sfortunatamente, se qualcuno volesse condurre la propria analisi sulle iniezioni di mRNA, lo farebbe non hanno un elenco adeguatamente dettagliato degli ingredienti con cui confrontarlo, né accesso alla metodologia normativa stabilita su come testarne adeguatamente la purezza.
È perché i produttori ed la FDA considera tutti gli ingredienti di queste iniezioni di mRNA, inclusa la sequenza dell'mRNA più le proprietà delle nanoparticelle lipidiche (LNP), tra cui emivita, strutture LNP, modificazioni superficiali, numero/tipo(i) di LNP per dose e punti di attacco su il filamento di mRNA, da non specificare o “segreto commerciale”.
Oltre a ciò, la FDA considera anche il metodologie anche su come testare la purezza delle iniezioni di mRNA è un segreto commerciale.
Supporto bipartisan e centinaia di miliardi di dollari dei contribuenti, ma NON trasparenza?
La segretezza sull’mRNA del Covid esiste anche se sia l’amministrazione Trump che quella Biden avevano proposto la piena trasparenza con le iniezioni di mRNA al punto da revocare i diritti di proprietà intellettuale sull’mRNA del Covid. Nonostante ciò, sia la FDA che i produttori stanno consentendo/mantenendo uno stretto controllo sui brevetti, compresi i dati di base su questi vaccini, come segreto commerciale. Lo stanno facendo nonostante tutti i produttori di vaccini anti-Covid lo abbiano ricevuto centinaia di milioni di dollari dei contribuenti secondo Forbes/Statista pubblicazioni.
Studiare l’epidemiologia della sicurezza dei farmaci è già abbastanza difficile. Senza la purezza/coerenza verificabile del prodotto, una valutazione completa della sicurezza è impossibile.
La piena trasparenza di tutti gli ingredienti e le misure di controllo della qualità sono importanti non solo perché sono state pesantemente finanziate dai contribuenti con centinaia di milioni di dollari, ma perché sono sorte una serie di domande sulla sicurezza e sull’efficacia delle iniezioni di mRNA di Covid.
Oltre ad essere eccezionalmente complessa, la loro approvazione è stata successivamente accelerata dalle autorità di regolamentazione meno di un anno. La maggior parte dei farmaci e dei vaccini in genere vengono portati in giro dieci anni per testare completamente la sicurezza/efficacia e rivederlo e approvarlo. Oltre al fatto che gli ingredienti sono completamente nuovi, molto complessi e i primi nel loro genere ad essere somministrati su larga scala, lo sviluppo include le valutazioni cliniche a lungo termine sulla sicurezza/tossicità e le revisioni epidemiologiche sono state accelerate e probabilmente non completamente chiarite prima del rilascio.
La verifica, la trasparenza e la “veridicità” degli ingredienti della FDA hanno precedenti che risalgono al 1800:
La verifica analitica e la trasparenza degli ingredienti ovvero la “verità nell'etichettatura” dove si trova il contenuto della bottiglia necessario per abbinare gli ingredienti elencati è antecedente alla fondazione della FDA, nel 1862. La FDA di oggi è in realtà nata da ciò che era iniziato come un singolo dipendente del “Dipartimento di Chimica” impiegato presso il Dipartimento dell’Agricoltura degli Stati Uniti.
Adulterazione, (ingredienti alterati o tossici) marchio errato (contiene un'etichetta falsa o è altrimenti fuorviante o contiene indicazioni mediche errate), o etichettatura errata (contiene uno o più ingredienti non elencati sull'etichetta del prodotto) hanno tutti avuto una lunga e brutta storia in America. Si pensava che l’eccesso avesse raggiunto il picco nella prima metà del XIX secolo – o almeno in quel periodo divenne identificabile – poiché solo nel 19 erano stati sviluppati processi tecnici per analizzare e rilevare le frodi sugli ingredienti. Prima di ciò, i cosiddetti “stregoni viaggianti” che si autodefinivano “dottori” (invariabilmente con credenziali dubbie o inesistenti) spacciavano flaconi di prodotti “panaceutici”, le cui etichette degli ingredienti elencavano solo contenuti nebulosi o innocui come “vitamine""estratti di erbe," o "olio di serpente” – o spesso non hanno alcuna lista degli ingredienti.
Allora, molti devoti e puritani del New England, che per motivi religiosi lo avrebbero fatto mai toccare l'alcol, acquisterebbero queste soluzioni da questi venditori ambulanti e verrebbero inconsapevolmente indotti a consumare soluzioni che non solo contenevano alcol, ma narcotici come oppio e/o cocaina. Con la scusa di migliorare una cornucopia assurdamente ampia di disturbi, i pazienti svilupparono invece una dipendenza punitiva e/o subirono un impatto negativo sulla loro salute da questi primi “spacciatori di droga”.
Man mano che il problema cresceva, il governo federale iniziò a prenderne atto. Alla fine, il Pure Food and Drug Act fu approvata nel 1906 e portò alla creazione della Food and Drug Administration (FDA).
[La FDA aveva una formativa dovere di garantire che i farmaci riportino dichiarazioni di etichettatura veritiere e soddisfino determinati standard di purezza e forza.
Ricorda che ha quasi 120 anni requisito di etichettatura veritiera e la parte "purezza" del Pure Food and Drug Act del 1906 mentre leggi sui test di verifica dell'mRNA e sulla trasparenza degli ingredienti.]
Quali test di verifica degli ingredienti “veritieri” e “puri” vengono eseguiti per i prodotti regolamentati dalla FDA?
Nel 2021, la FDA ha deciso di iniziare a monitorare la qualità farmaceutica americana tramite a raccolta a distanza of invio tramite posta dei campioni per i farmaci in sostituzione delle ispezioni nelle strutture vive a causa della pandemia di Covid. Era legale? Potrebbe mai essere considerato scientificamente appropriato? Oggi, nonostante la fine della pandemia, è in corso l’unico test ufficiale sul rilascio dei farmaci in qualsiasi Farmaceutico Covid mRNA appare a ancora essere effettuato dalla FDA tramite un fornitore fornito dal produttore, "spedito per posta” campione secondo a screenshot dell'attuale sito web della FDA. Ovviamente, un metodo di campionamento “inviato per posta” è molto diverso e potenzialmente meno affidabile rispetto alla raccolta diretta dei campioni tramite un metodo di raccolta diretta di persona. Nonostante ciò, la FDA afferma di aver “lo standard più elevato in tutto il mondo per il campionamento e i test. "
Inoltre, la FDA propone di promuovere ulteriormente la sua politica di test remoti “inviati tramite posta” con un documento orientativo appena proposto.
Sebbene esista solo come “bozza” di documento FDA, i siti web ufficiali della FDA lo mostrano l'invio dei campioni sembra essere già stato implementato almeno da gennaio 2021. Sembra che la FDA affermi i risultati di questi test spediti per posta come verifica indipendente.
Inoltre, il fondo della prima pagina della bozza della FDA Il documento propone l’espansione dei “test a distanza”. Attualmente elenca ogni Divisione di regolamentazione dei prodotti della FDA presso la FDA, il che implica che si tratta di una proposta politica a livello di agenzia.
L'elenco completo comprende:
- Ufficio Affari Regolatori
- Ufficio per la politica e la risposta alimentare
- Ufficio dei prodotti combinati
- Centro per la valutazione e la ricerca biologica
- Centro per la valutazione e la ricerca sui farmaci
- Centro per i dispositivi e la salute radiologica
- Centro per la sicurezza alimentare e la nutrizione applicata
- Centro per i prodotti del tabacco
- Centro di Medicina Veterinaria
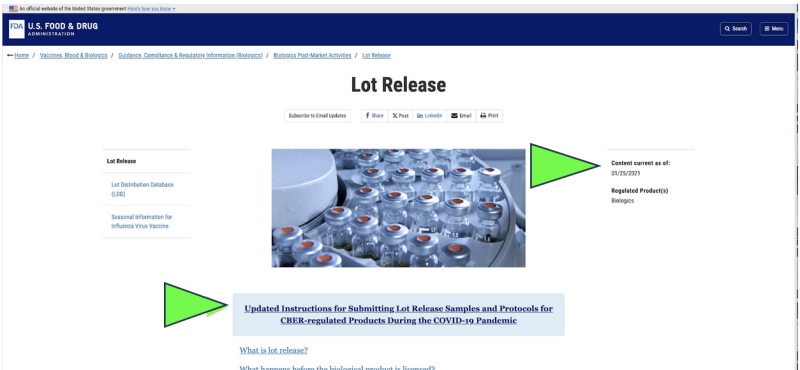
Il campionamento del controllo di qualità "inviato per posta" da parte della FDA è appropriato? Cosa accadrebbe se le ispezioni dei ristoranti del Dipartimento sanitario statale rispecchiassero la politica della FDA?
Questa metodologia di campionamento "per posta" è altrettanto assurda, ad esempio, nel caso in cui il dipartimento sanitario di uno stato monitori i ristoranti chiedendo loro di "inviare periodicamente per posta" vari elementi dal loro menu a una struttura di analisi in modo che i dipartimenti sanitari possano testare potenziali alimenti contaminazione nata e/o chiedere ai ristoranti di promettere di testare essi stessi le voci del menu. E se quel ristorante fosse in Cina? E se quel ristorante fosse in India? O qualsiasi altro paese noto per avere un storia abissale di frode e controllo di qualità i problemi?
Questa metodologia sarebbe inaccettabile sia per i ristoranti che per le aziende farmaceutiche, per ragioni che includono l’ovvio: i produttori potrebbero inviare i campioni che preferiscono, non necessariamente campioni di lotti rappresentativi. Ovviamente non è la stessa cosa che gli ispettori della FDA acquisiscono campioni durante ispezioni senza preavviso dell’intera struttura.
Seguendo l'analogia con il ristorante, ovviamente tutti i ristoranti lo farebbero inviare campioni di grado “A”. che non sarebbe necessariamente rappresentativo di ciò che ricevono i consumatori.

Controllo di qualità: cos'è il "test di rilascio" farmaceutico e perché è importante?
Oggi, la FDA supervisiona la qualità e il contenuto di $2.7 trilioni valore del prodotto ogni anno, ma sembra che stia sopprimendo le valutazioni e i risultati critici della verifica degli ingredienti. La FDA dovrebbe proteggere gli americani conducendo globale test analitici come checksum per garantire l'accuratezza degli ingredienti. I risultati di ciò dovrebbero essere trasparenti per i contribuenti che finanziano il I 6.6 miliardi di dollari della FDA bilancio. Quella verifica scientifica si chiama farmaceutica”test di rilascio.” Test di rilascio è un termine tecnico che si riferisce a un processo che coinvolge una varietà di analisi strumentali utilizzate completo testare i prodotti per purezza, concentrazione, consistenza, identità e impurità di qualsiasi tipo.
L’intera FDA è nata da quel dipendente del “Dipartimento di Chimica” del 1862 e dalla necessità di trasparenza e verifica degli ingredienti. Oggi, quel dipendente è proliferato in un intero dipartimento della FDA composto da 1,300 scienziati e personale di supporto presumibilmente dedicato alla verifica degli ingredienti tramite test di rilascio farmaceutico. Quelli della FDA Ufficio di Qualità Farmaceutica (OPQ) dovrebbe garantire che i prodotti farmaceutici corrispondano esattamente al contenuto degli ingredienti elencati, senza variabilità di qualità/impurità (qualitativa) o contenuto (qualitativa). Le norme che lo richiedono sono molto specifiche e dettagliate 21 CFR § 201.10.
Come la FDA verifica le iniezioni di mRNA per il controllo di qualità:
I risultati del controllo di qualità dei test delle iniezioni di mRNA erano particolarmente critici perché erano grandi, complessi e venivano realizzati rapidamente. Mentre i contribuenti dipendono dalla FDA per verificare la qualità dell’iniezione di mRNA e condividere i risultati, la FDA sembra obbligati a proteggere gli ingredienti dei produttori a scapito anche della più elementare trasparenza riguardo ai prodotti mRNA Covid. Anche se sembra che la FDA raccolga campioni, la loro metodologia di “invio tramite posta” è fondamentalmente errata. Inoltre, la FDA non condivide i risultati di tali test da nessuna parte in cui potrei trovarli.
In altre parole: durante la pandemia, quando le iniezioni di mRNA nuove di zecca e ampiamente implementate venivano imposte agli americani a “velocità di curvatura” e quando l’America faceva più affidamento sugli obblighi di qualità/normativi della FDA, la FDA accettava “inviati per posta” auto-presentati. in” test e/o risultati del controllo qualità. La FDA non lo ha considerato? I produttori di mRNA hanno ammesso di “faticare[d]” per rispondere alla produzione e di “fare fatica” per tenere il passo con i processi produttivi? I produttori degli ingredienti dell’mRNA hanno inoltre affermato che gli sforzi per soddisfare le esigenze erano “senza precedenti”.
Dichiarazioni come questa non danno fiducia ai consumatori nella qualità e sono illustrative di un enorme miglioramento di questi prodotti complessi che dovrebbe garantire particolarmente vigile e il controllo di persona della FDA su strutture e prodotti fabbricati, pandemici o meno. Un produttore di ingredienti per l’mRNA, ad esempio, ha dichiarato di aver improvvisamente aumentato la propria produzione del 50 volte.
Nel mezzo di quella nuova tecnologia spinta avanti a “velocità di curvatura”, nessuno dei 1,300 scienziati dell’OPQ presso la FDA richiedeva ispezioni dal vivo, o almeno si offriva di fare altro che chiedere campioni “inviati per posta” potenzialmente discutibili per i test?
La domanda ovvia è: perché la FDA non ha raccolto direttamente i campioni? Anche con la pandemia in atto, la FDA avrebbe potuto ispezionare strutture che indossavano tute ignifughe o – o al molto almeno – ha scelto di raccogliere campioni da farmacie, ospedali o magazzini dei distributori.
Metodologia nascosta per testare gli ingredienti dell'iniezione di mRNA:
Al di là dell’assenza di risultati dei test e di discutibili risultati di campionamento “inviati per posta”, la FDA lo è inoltre nascondendo la loro metodologia convalidata impedendo ad altri di eseguire analisi proprie e indipendenti sulla qualità/purezza delle iniezioni di mRNA.
Analizzare in modo indipendente la purezza e la potenziale contaminazione dei farmaci rispetto alla lista degli ingredienti è qualcosa che avevo tentato di fare io stesso quando ho concettualizzato il primo farmacia analitica. Tuttavia, poiché le iniezioni di mRNA sono una tecnologia innovativa con un elenco di ingredienti tutt’altro che del tutto trasparente, la metodologia di test che sarebbe necessario impiegare non è semplice come lo sarebbe per altri farmaci a piccole molecole. Chiunque cerchi di verificare la conservazione, la stabilità, la specificità, la chimica, la sensibilità o anche la metodologia di base per la validazione dei test e/o i risultati viene bloccato da un rapporto della FDA contenente revisioni ridicolmente invasive, che impediscono anche la comprensione scientifica più fondamentale di come valutare potenzialmente risultati o condurre test impossibili.
Come esempio visivo toccante, una singola pagina oscurata in un riepilogo normativo della FDA più lungo (mostrato di seguito) fa parte di un 127-documento di pagina (di cui solo 63 pagine sono state condivise e di queste 63 pagine circa il 50% è stato oscurato) su come valutare la purezza, la concentrazione e altre misure analitiche delle iniezioni di mRNA.
Quelli Redazioni FDA (b)(4). specificate redazioni dettagliate utilizzate per “proteggere i segreti commerciali e le informazioni commerciali o finanziarie riservate.” Ma è davvero appropriato etichettarlo come “commerciale” se la ricerca/sviluppo/prodotto è stato finanziato con? centinaia di milioni di dollari dei contribuenti?

Senza un elenco degli ingredienti o una metodologia di analisi, è impossibile per chiunque altro al di fuori della FDA o dei produttori sapere esattamente come verificare la presenza del prodotto. adulterazione (ingredienti alterati o tossici) o etichettatura errata (perché un elenco completo degli ingredienti inclusa la sequenza nucleotidica e il configurazioni di nanoparticelle lipidiche sono particolarmente vaghi sull’etichetta del prodotto).
La mancanza di metodologia è particolarmente problematica poiché ne hanno dato prova nuovi dati preliminari che utilizzano una metodologia indipendente Contaminazione del DNA nelle iniezioni di mRNA Covid.
Quindi, se un individuo esterno affermasse di aver testato e trovato un'impurità nelle iniezioni di mRNA e chiedesse alla FDA o ai produttori la sua risposta, verrebbe accolto con una risposta che afferma qualcosa sulla falsariga di:
- Non hai utilizzato una metodologia di test convalidata/appropriata per giungere alle tue conclusioni e pertanto le tue analisi non sono valide.
Per questo, il laboratorio indipendente tenterà di richiedere la metodologia di test dalla documentazione approvata dalla FDA (ovvero, il documento completo contenente figura 5) chiedendo: “Va bene, vorrei testarlo utilizzando la metodologia da voi approvata; ci dirai di cosa si tratta?"
- La FDA o il produttore risponderebbero qualcosa del tipo: “Ciò che siamo disposti a divulgare sulla metodologia impiegata che non sia riservata può essere trovato online o tramite una richiesta FOIA della FDA” …dove si sarebbero incontrati il seguente documento pesantemente oscurato, dove qualsiasi cosa anche lontanamente significativa è ricoperta da redazioni (b)(4).
Leggere tra le righe: è ovvio che sia i produttori che la FDA americana non vogliono che nessun altro oltre a loro conosca gli ingredienti completi o addirittura testi le iniezioni di mRNA per verificarne la purezza e la consistenza.
Secondo i funzionari della FDA: la produzione farmaceutica lo è Altamente Incline all'errore:
Molti le cose possono – e vanno – andare storte durante il processo di produzione farmaceutica. Oltre alle potenziali incoerenze con le iniezioni di mRNA/LNP, implicano problemi qualitativi e quantitativi ogni Prodotto farmaceutico regolamentato dalla FDA. Anche la Camera e il Senato hanno formalmente riconosciuto le denunce relative al fallimento della FDA nel garantire la catena di fornitura farmaceutica americana. La maggioranza dei Il settore farmaceutico americano prodotto per l'utente finale di consumoviene prodotto all’estero in paesi come India e Cina, e altri paesi a basso costo di manodopera lo sono non ben considerato per gli alti livelli di controllo di qualità. Il registro federale è pieno di segnalazioni di violazioni negli stabilimenti produttivi indiani e cinesi.
La FDA sta certificando anche questi stabilimenti, compresi quelli con una lunga storia di violazioni, tramite un sistema di “invio per posta” alla FDA? Stranamente, la risposta alla domanda è qualcosa che metterebbe molto a disagio chiunque sia interessato alla qualità farmaceutica.
Mentre un Six Sigma Il livello di precisione è stato a lungo l'obiettivo per la qualità e la sicurezza nel settore automobilistico, dei computer, della telefonia mobile e di altri prodotti ad alta tecnologia, sembra essere stato per lo più trascurato quando si tratta della produzione farmaceutica.
I funzionari della FDA hanno pubblicato dati che stimano un'imprecisione di 2-3σ (sigma) nella produzione farmaceutica. Una qualità 2σ corrisponde a 308,537 difetti per 1,000,000 di opportunità. (Ci sono probabilmente più di 1,000,000 di possibilità di errore quando si tratta di produzione farmaceutica.) La FDA ne è consapevole ai più alti livelli di leadership; infatti, la corrente Il capo dell’Ufficio di qualità farmaceutica della FDA, Michael Kopcha ha persino scritto e pubblicato il suddetto calcolo Six Sigma, lamentando la natura imprecisa della produzione farmaceutica indietro nel 2017.
La latitudine di errore per i prodotti mRNA e/o i loro LNP potrebbe essere pari meno precisi rispetto a 2-3σ (più basso è σ, più errato è un prodotto) poiché includono materiale nucleotidico e nuovi LNP, rendendoli sostanzialmente più complessi dei prodotti farmaceutici a piccole molecole, nonostante siano stati sviluppati, prodotti e rilasciati a " velocità di curvatura."
Anche se la FDA e i suoi funzionari riconoscono un’imprecisione intrinseca nella produzione, perché nel vasto mondo dello sport la FDA non sta adempiendo alla sua missione di sicurezza condividendo pubblicamente i test di rilascio della tecnologia mRNA con il pubblico americano che la finanzia?
Ancora prima del 1862? Le iniezioni di mRNA sono gli unici farmaci per i quali gli americani non hanno? Completato Informazioni sugli ingredienti?
La mancanza di chiarezza sul numero di sequenze di mRNA e altre informazioni critiche è in diretto contrasto con un altro farmaco a base di RNA approvato dalla FDA: patisiran (Onpattro®). Onpattro fornisce in modo trasparente la sequenza, il peso molecolare e la concentrazione in milligrammi dei suoi prodotti all'interno della FDA ufficiale etichettatura della confezione come illustrato in un estratto qui sotto:


Mancanza di mRNA Covid Specificità della dose: 0.3 ml (o 0.5 ml) di cosa?
Al momento, non disponiamo ancora di informazioni di base sugli ingredienti di alcuna iniezione di mRNA Covid. I farmacisti sanno solo dare una specifica volume di fluido, e apparentemente lo fece senza fare domande. Normalmente, l’etichettatura ufficiale della confezione della FDA dovrebbe dettagliare gli ingredienti effettivi in quel volume, ma non per le etichette dell’mRNA Covid: indicano semplicemente 0.3 ml (o 0.5 ml) come “Forma di dosaggio e concentrazione”.
Inoltre, come potrebbe dirti qualsiasi studente delle scuole superiori, 0.3/0.5 ml è un volume, non a forza. Non conosciamo alcuna specifica quantitativa di ciò che è contenuto in quegli 0.3/0.5 ml come ad esempio: quante particelle LNP? Quali dimensioni/morfologie di questi LNP? Quante sequenze di mRNA in quel volume?




È questo ciò che viene considerato adeguatamente trasparente o “etichettatura veritiera” dalla FDA?
L'estratto taglia e incolla del foglietto illustrativo sopra riportato contiene tutte le informazioni che i produttori condividono con i consumatori riguardo alla dose - che è tristemente inadeguata rispetto a tutte le altre etichette della FDA - o chiunque sia curioso di sapere qualcosa oltre alla quantità di fluido da iniettare e la concentrazione di 30 o 100 mcg di una sequenza di mRNA non specificata.
La notevole imprecisione di questa etichetta consentita dalla FDA sembra essere in conflitto in particolare con la sua etichetta di quasi 120 anni: “richiedendo che alimenti e farmaci rechino dichiarazioni veritiere sull'etichetta e soddisfino determinati standard di purezza e forza. "
È questo quello che viene considerato un elenco “veritiero” di ingredienti dalla FDA? (Vedere 21CFR §352e 21 CFR §201.10 relativi alla “dichiarazione degli ingredienti” e ai “farmaci e dispositivi con marchio errato”).
La domanda è: elenca ingredienti sconosciuti o non specifici che nessuno tranne il produttore può decifrare veramente soddisfare lo spirito o i requisiti legali di “etichettatura”? Quell’etichetta è considerata “veritiera” dalla FDA americana? In ogni caso, da che parte sta la FDA? produttori o consumatori?
Oltre a non essere specificato direttamente, il numero esatto di filamenti di LNP o di mRNA in un’iniezione da 30 o 100 mcg non può nemmeno essere estrapolato stechiometricamente o sulla base di Il numero di Avogadro, perché la sequenza dell'mRNA, il peso molecolare e/o i componenti/configurazioni dell'LNP non sono forniti da nessuna parte nell'etichettatura ufficiale della FDA.
Come si può sapere se il numero di filamenti di mRNA per codificare la proteina spike per Covid è proporzionale al carico di inoculo Covid che si riceverebbe da un’infezione acquisita in comunità? Risposta: non possono.
Sono iniezioni di mRNA Covid Etichettato in modo appropriato/etichettato erroneamente?
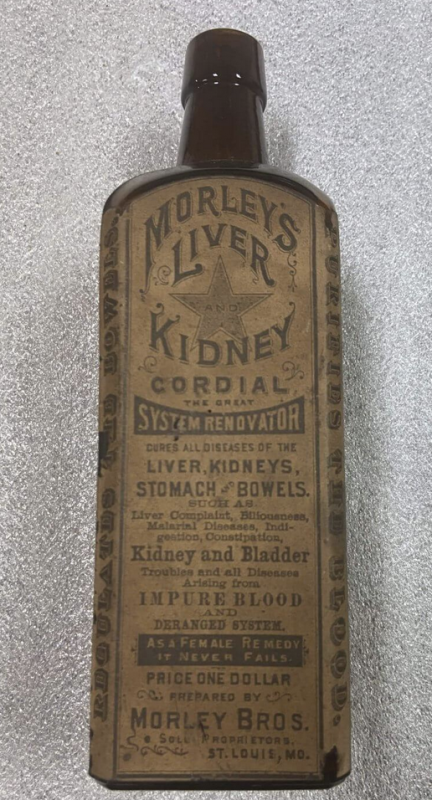
21 CFR 211.125 specifica “Dovrà essere esercitato uno stretto controllo sulle etichette rilasciate per l’uso nelle operazioni di etichettatura dei prodotti farmaceutici,” ma sembra che la FDA sia stata così negligente con l’etichettatura approvata delle iniezioni di mRNA Covid nonostante ciò ogni altro farmaco – incluso Onpattro basato su mRNA – specifica tale informazione. Storicamente, le decisioni normative della FDA (come quali informazioni includere nell’etichettatura del prodotto) si basano sulla precedenza e le iniezioni di mRNA Covid rappresentavano un’ovvia deviazione dalla precedenza storica e legale della FDA. Quella notevole assenza di dati e mancanza di chiarezza ricorda i tempi del Cordiale per fegato e reni di Morley alla fine del 1800. La differenza è: allora la FDA non esisteva, ma oggi esiste una FDA con circa 20,000 dipendenti, almeno alcuni dei quali apparentemente credevano che questa etichetta fosse trasparente e “veritiera”.
Dichiarare un ingrediente sconosciuto/indecifrabile/oscuro che nessuno avrebbe mai potuto determinare con precisione probabilmente non è ciò che intendevano i legislatori del Pure Food and Drug Act del 1906 quando specificarono le regole della FDA sulla “etichettatura veritiera”. A parte questo: il fatto che le dosi sono raddoppiate per volume di diversi produttori (30 mcg/0.3 ml vs 100 mcg/0.5 ml) significa che queste sequenze di mRNA sembrano essere molto diverse nella lunghezza dei nucleotidi e, a loro volta, lo avrebbero fatto più e diversi LNP più allegati. Ciò significa che le sequenze di mRNA utilizzate per trascrivere la proteina spike hanno dimensioni circa doppie (10 mcg/0.1 ml contro 20 mcg/0.1 ml) rispetto a quelle dei diversi produttori, o c'è qualcos'altro che contribuisce alla differenza di lunghezza del nucleotide?
Per i non addetti ai lavori che stanno ancora leggendo fino a questo punto (complimenti, a proposito): la mancanza di informazioni dettagliate sull'etichettatura potrebbe essere come pubblicizzare in modo generico una casa in vendita, affermando che è fatta di legno e mattoni, su una lastra di cemento - ma senza mostrare eventuali immagini della casa (ad es. sequenza) e che non ne condividano la metratura (ad es. peso molecolare). In ogni caso, la mancanza di informazioni è inadeguata e rappresenta una deviazione dagli standard tradizionali.
Ogni altro farmaco approvato dalla FDA, compresi altri farmaci mRNA, contiene informazioni complete sugli ingredienti sui loro prodotti, incluso una rappresentazione strutturale e un peso molecolare del loro prodotto in modo che le persone sappiano esattamente cosa stanno ottenendo.
È vero: cerca qualunque farmaco ti venga in mente nel Banca dati Drugs.com e notate come tutte le etichette forniscono struttura e/o peso molecolare. La prova che i colpi di mRNA di Covid sono a cospicuo eccezione alla storica pratica di approvazione della FDA e alla regola dell'"etichetta veritiera".
Uno studio danese del 2023 descrive in dettaglio la variabilità clinica significativa tra lotti di iniezioni di mRNA di mRNA Covid-19:
La mancanza di trasparenza anche sulla validazione dei test “inviati per posta” potenzialmente non validi sembra aver dato ai produttori un passaggio su un’altra parte di fondamentale importanza di ciò che la FDA supervisiona: potenziali manifestazioni cliniche sulle variazioni di lotto/lotto di iniezioni di mRNA. Una retrospettiva Studio danese sulla sicurezza pubblicato all'inizio del 2023 ha dettagliato un modello altamente deviante di segnalazioni di eventi avversi dalle iniezioni di mRNA BNT162b2 di Pfizer-BioNTech in correlazione con il sistema danese di segnalazione di eventi avversi DKMA.
Nel grafico a linee che segue, diversi punti colorati rappresentano diversi lotti di iniezioni di mRNA di Pfizer-BioNTech. Ha separato i lotti in tre diverse categorie; numero da alto-basso a (quasi) assente di gruppi di eventi avversi segnalati (rispettivamente grafici blu, verde e giallo).
In altre parole: prodotti presumibilmente “equivalenti” dello stesso produttore sembrano avere incidenze estremamente diverse di eventi avversi, a seconda del lotto, con ciascuno di questi lotti che rappresenta centinaia di migliaia di iniezioni di mRNA.
Quando sono state aggiunte le corrispondenti linee di regressione lineare, è emerso uno schema particolare:
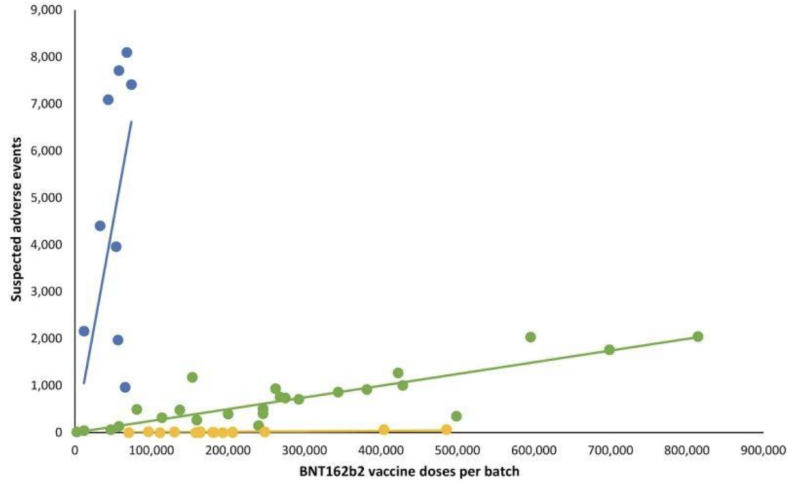
Domande importanti sulla notevole disparità degli eventi avversi tra i lotti di mRNA di Covid-19 includono:
- Le variazioni degli eventi avversi potrebbero essere dovute a variazioni qualitative o quantitative nelle sequenze di mRNA o nel numero di filamenti di mRNA tra i lotti?
- Le variazioni degli eventi avversi potrebbero essere dovute a variazioni qualitative o quantitative nelle dimensioni/morfologie o nella quantità di LNP tra i lotti? A quali test sono stati eseguiti garantire la sicurezza dei vari LNP usato nelle iniezioni di mRNA?
- Quei lotti che corrispondevano ai punti dati giallo rispetto a quello verde rispetto a quello blu erano in qualche modo qualitativamente o quantitativamente diversi?
- Lo stoccaggio/la movimentazione post-produzione è stato compromesso presso la struttura di somministrazione (o in qualche altro punto lungo la catena di fornitura) portando alla variabilità del prodotto?
- Qual è il tasso di errore/Sigma di questo e di altri prodotti provenienti da un particolare impianto di produzione/capoturno responsabile della produzione?
- Gli ingredienti di questi prodotti Covid mRNA provenivano dall’India o dalla Cina e non altrove, a seconda del lotto?
- Quali percentuali di lotti di prodotti Covid mRNA sono stati testati tramite ritiro di persona da un ispettore della FDA rispetto a quelli "spediti" dall'inizio ad oggi? Ogni singolo lotto è stato testato utilizzando solo uno di questi due metodi di raccolta?
- La FDA ha eseguito la verifica dei test di rilascio sui lotti del sistema danese di segnalazione degli eventi avversi DKMA? Se sì, perché la FDA non rilascia quei particolari risultati dei test? In caso contrario, perché non sono stati effettuati i test?
- Esiste un problema fondamentale nel produrre in modo coerente LNP e/o sequenze di mRNA in modo affidabile e senza contaminazione?
I risultati dello studio danese e le domande di cui sopra sugli eventi avversi potrebbero *iniziare* ad essere affrontati, ma non senza che la FDA condivida in modo indipendente i risultati dei test di rilascio. Allo stato attuale, a causa delle onnipresenti redazioni della FDA (b) (4), nessuno conosce la metodologia convalidata per testare i colpi di mRNA di Covid or esattamente quali lotti nello studio danese sono stati o non sono stati testati or i risultati di tali test batch.
…Anche se la FDA avesse scelto di rilasciare i risultati dei test sui lotti, come fanno i consumatori a sapere se tali risultati sono rappresentativi dei lotti specificati, dal momento che i produttori selezionano autonomamente quali campioni “spedire”?
Non garantire la trasparenza degli ingredienti e non garantire la qualità tramite una metodologia di campionamento adeguata è un requisito fondamentale e basilare della FDA. In effetti, è stata la ragione principale per la formazione della FDA! Gli americani non meritano forse migliori leggi in materia di trasparenza, supervisione e “etichettatura veritiera” quando si tratta dei nostri prodotti farmaceutici, soprattutto da quando tali leggi sono state emanate oltre 100 anni fa?
Pubblicato sotto a Licenza internazionale Creative Commons Attribution 4.0
Per le ristampe, reimpostare il collegamento canonico all'originale Istituto di arenaria Articolo e Autore.








