
In prima parte di questo articolo, ho esaminato il quadro contrattuale e normativo applicato dal governo degli Stati Uniti allo sviluppo iniziale, alla produzione e all’acquisizione delle iniezioni di mRNA Covid, utilizzando gli accordi BioNTech/Pfizer per illustrare il processo.
Ho dimostrato che a questi prodotti è stata concessa l'autorizzazione all'uso di emergenza (EUA) sulla base di studi clinici e processi di produzione condotti con
- nessuna norma giuridica vincolante,
- nessuna supervisione o regolamentazione della sicurezza legalmente vietata e
- nessun risarcimento legale da parte del produttore per potenziali danni.
In questo articolo di follow-up, fornirò un’analisi dettagliata della documentazione sottostante.
Other Transaction Authority/Agreement (OTA): un percorso di acquisizione militare
I accordo tra il governo degli Stati Uniti, rappresentato dal Dipartimento della Difesa (DoD), e Pfizer, in rappresentanza della partnership BioNTech/Pfizer, nel luglio 2020, per l’acquisto di un “vaccino per prevenire il COVID-19” non era un normale contratto di acquisizione.
Si trattava di un accordo ai sensi dell'Other Transaction Authority (OTA) - un percorso di acquisizione che, secondo Linee guida del Dipartimento della Difesa, è stato utilizzato dal 1958 per “permettere ad un'agenzia federale di stipulare transazioni diverse da contratti, sovvenzioni o accordi di cooperazione. "
[GRESETTO AGGIUNTO]
Una revisione approfondita dell'uso dell'OTA da parte del DoD, inclusa la sua storia statutaria, è disponibile nel Rapporto del Servizio di ricerca del Congresso del 22 febbraio 2019. Questo rapporto, insieme a ogni altra discussione sulle OTA, specifica che si tratta di un percorso di acquisizione alternativo per scopi militari e di difesa. Non è destinato, né è mai stato utilizzato prima del Covid, per nulla destinato principalmente ad uso civile.
Se cerchi Leggi OTA nel Codice degli Stati Uniti, questo è il percorso che seguirai:
Forze Armate -> Diritto Militare Generale -> Acquisizioni -> Ricerca e Ingegneria -> Accordi -> Autorità del Dipartimento della Difesa per la realizzazione di alcuni progetti prototipo
Questo percorso legale mostra molto chiaramente che le leggi dell'OTA sono destinate all'acquisizione di prototipi di ricerca e ingegneria per le forze armate.
Secondo il sito web della DARPA,
Il Dipartimento della Difesa ha autorità su tre diversi tipi di OT: (1) OT di ricerca, (2) OT prototipo e (3) OT di produzione.
Questi tre tipi di OT rappresentano tre fasi di ricerca iniziale, sviluppo di un prototipo ed eventuale produzione.
All'interno di queste tre tipologie, ci sono categorie specifiche di progetti a cui l'OTA può candidarsi:
- Originariamente, secondo l' Panoramica dell'OTA fornita dal Dipartimento della Difesa, l'Altra Autorità per le Transazioni era "limitata ad applicarsi alle armi o ai sistemi d'arma proposti per essere acquisiti o sviluppati dal Dipartimento della Difesa".
- L'OTA è stata successivamente ampliata per includere "qualsiasi progetto prototipo direttamente correlato al miglioramento dell'efficacia della missione del personale militare e delle piattaforme, sistemi, componenti o materiali di supporto proposti per essere acquisiti o sviluppati dal Dipartimento della Difesa, o al miglioramento di piattaforme, sistemi, componenti o materiali in uso alle Forze Armate”.
Finora, nulla di tutto ciò sembra un percorso di acquisizione di milioni di nuovi prodotti medici destinati principalmente all’uso civile.
Esiste qualche eccezione per l’uso civile dell’OTA che potrebbe applicarsi ai vaccini Covid mRNA?
I Legge sull'autorizzazione della difesa nazionale per l'anno fiscale 2004 (PL 108-136) conteneva una sezione che conferiva altra autorità di transazione al "capo di un'agenzia esecutiva che si impegna in ricerca di base, ricerca applicata, ricerca avanzata e progetti di sviluppo" che "hanno il potenziale per facilitare la difesa contro o il recupero dal terrorismo o da attacchi nucleari, biologici, attacco chimico o radiologico”.
Questa disposizione è stata prorogata fino al 2018, ma non sembra essere stata prorogata oltre tale anno. Inoltre, si noti che anche in questo caso eccezionale di utilizzo dell'OTA da parte di soggetti non DoD, la situazione deve riguardare il terrorismo o un attacco con armi di distruzione di massa (CBRN).
Quali altre leggi sulle OTA potrebbero applicarsi?
Il rapporto CRS del 2019 sopra citato fornisce questo grafico, che mostra che alcune agenzie non DoD hanno alcune OTA o autorità correlate:
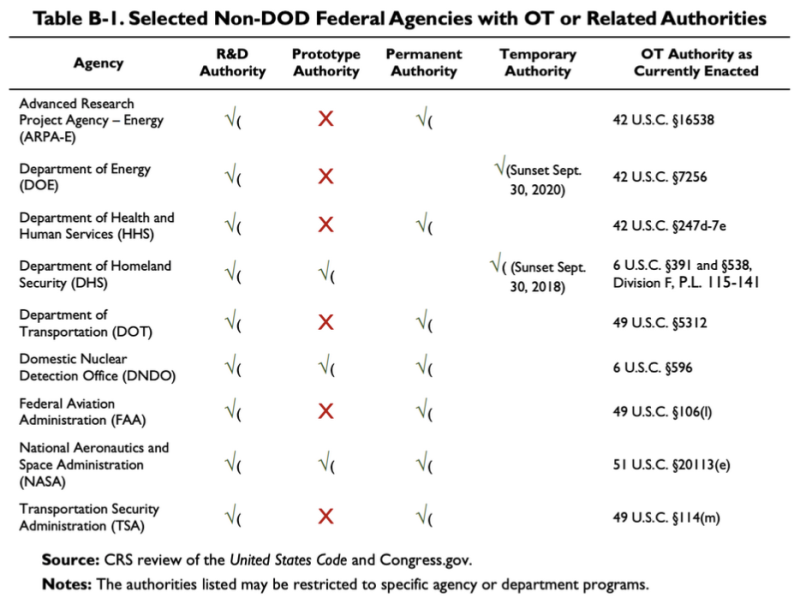
Secondo questa tabella, il Dipartimento della salute e dei servizi umani (HHS) ha alcune altre autorità di transazione di ricerca e sviluppo (R&S). La legge relativa al L'autorità OT dell'HHS è 42 U.S.C. §247d-7e.
Dove si trova questa legge e cosa dice?
Sanità e welfare pubblico -> Servizio sanitario pubblico -> Poteri e doveri generali -> Cooperazione tra Stati federali -> Autorità per la ricerca e lo sviluppo avanzato biomedico (BARDA) -> Autorità di transazione
Quindi c’è un punto nella legge relativa alla salute e al welfare civile in cui l’OTA potrebbe essere applicabile, sebbene sia valida solo per ricerca e sviluppo, non per prototipi o produzione.
La legge afferma che il segretario della BARDA ha Autorità OT
rispetto ad un prodotto che è o potrebbe diventare a contromisura qualificata o prodotto qualificato pandemico o epidemico, attività che prevalentemente:
(i) sono condotti dopo la ricerca di base e lo sviluppo preclinico del prodotto; E
(ii) siano legati alla fabbricazione del prodotto su scala commerciale e in una forma che soddisfi i requisiti normativi previsti dalla Federal Food, Drug and Cosmetic Act [21 USC 301 e segg.] o sotto sezione 262 di questo titolo.
[GRESETTO AGGIUNTO]
I “requisiti normativi” elencati nella legge significano che sarebbe impossibile per BARDA/HHS stipulare accordi – anche solo di ricerca e sviluppo – per qualsiasi prodotto medico (come i vaccini a mRNA) che non sia stato sottoposto a rigorosi test di sicurezza e a una rigorosa supervisione della produzione.
La "partnership" dell'HHS con il Dipartimento della Difesa ha eluso le leggi sulla protezione civile
Per riassumere la situazione di Other Transaction Authority/Agreements rispetto alle autorità civili, in generale, e ai vaccini Covid mRNA, in particolare:
- L’OTA è stata scritta e codificata come un modo per i militari di acquisire armi e altri sistemi e attrezzature necessari senza molta burocrazia. Copre ricerca e sviluppo, prototipi e successiva produzione.
- L'unica OTA per un'agenzia sanitaria pubblica è per l'HHS e copre solo la ricerca e lo sviluppo, non i prototipi o la produzione.
- Anche l’OTA di ricerca e sviluppo data all’HHS richiede ancora che i prodotti siano fabbricati “in una forma che soddisfi i requisiti normativi” per la sicurezza di farmaci e vaccini.
In altre parole: non è possibile che HHS abbia potuto utilizzare le sue limitate OTA per firmare contratti per centinaia di milioni di nuovi prodotti medici.
Allora cosa ha fatto l'HHS?
Come ha osservato il Government Accountability Office (GAO). Rapporto di luglio 2021 sulla “Contrazione Covid-19”: L'HHS ha "collaborato" con il Dipartimento della Difesa per "sfruttare le autorità OTA del Dipartimento della Difesa... cosa che mancava all'HHS". (Pag. 24)
Quali sono le autorità OT del Dipartimento della Difesa per i prodotti medici?
Come discusso, l’OTA ha lo scopo di aiutare i militari a ottenere attrezzature e tecnologia senza troppi problemi burocratici. Nessuna delle leggi originali relative all’OTA menzionava altro che “piattaforme, sistemi, componenti o materiali” destinati a “migliorare l’efficacia della missione del personale militare”.
Ma cinque anni prima del Covid venne introdotto un utilizzo eccezionale delle OTA:
Nel 2015, DoD ha annunciato la creazione del CBRN Medical Countermeasure Consortium, il cui scopo era quello di utilizzare il percorso di acquisizione dell’OTA per “collaborare con il Dipartimento della Difesa per sviluppare contromisure mediche chimiche, biologiche, radiologiche e nucleari con licenza FDA”. [FDA = Food & Drug Administration]
Come descritto nell’annuncio del 2015, ciò includeva “tecnologie prototipo per contromisure mediche terapeutiche mirate a bersagli di tossine virali, batteriche e biologiche di interesse per il Dipartimento della Difesa”. L’elenco degli agenti includeva i principali agenti patogeni della guerra biologica, come l’antrace, l’ebola e il marburg.
L’annuncio prosegue specificando che “le tecnologie abilitanti possono includere modelli animali di malattie e patogenesi da tossine virali, batteriche o biologiche (vie multiple di esposizione), test, tecnologie diagnostiche o altre tecnologie di piattaforma che possono essere applicate allo sviluppo di MCM approvati o autorizzati”. [contromisure mediche].”
Anche se questo non assomiglia ancora alla produzione di 100 milioni di nuovi vaccini per uso civile, fornisce più margine di manovra per l’OTA rispetto alla molto limitata Other Transaction Authority concessa a HHS.
Mentre l’OTA dell’HHS richiede il rispetto di ampie normative in materia di sviluppo e produzione, il percorso dell’OTA affinché il Dipartimento della Difesa sviluppi contromisure mediche richiede solo la “licenza della FDA”.
Pertanto, utilizzando le altre autorità di transazione del Dipartimento della Difesa, sarebbe teoricamente possibile aggirare qualsiasi normativa di sicurezza, a seconda dei requisiti per la licenza FDA di un prodotto generato da OTA. Come vedremo, nel caso dei vaccini Covid mRNA, è stata concessa l’autorizzazione all’uso di emergenza, senza richiedere alcuna supervisione legale sulla sicurezza.
Autorizzazione all'uso di emergenza (EUA)
Ecco come funziona la Food & Drug Administration (FDA) descrive i suoi poteri EUA:
Sezione 564 della legge FD&C (21 USC 360bbb-3) consente alla FDA di rafforzare la tutela della salute pubblica contro gli agenti biologici, chimici, nucleari e radiologici.
Con questa autorità EUA, la FDA può contribuire a garantire che le contromisure mediche possano essere utilizzate in caso di emergenza per diagnosticare, trattare o prevenire malattie o condizioni gravi o potenzialmente letali causate da agenti biologici, chimici, nucleari o radiologici quando non esistono adeguate misure approvate. e le alternative disponibili (tra gli altri criteri).
È estremamente importante comprendere che questi poteri dell’EUA sono stati concessi nel 2004 in circostanze molto specifiche legate alla preparazione agli attacchi con armi di distruzione di massa, altrimenti note come agenti CBRN (chimici, biologici, radiologici, nucleari).
Come spiegato nella fattura sanitaria della legge di Harvard,
Alla fine, è stata la guerra al terrorismo a dare origine all’autorizzazione all’uso di emergenza. Dopo gli eventi dell'11 settembre 2001 e i successivi attacchi postali all'antrace, il Congresso ha promulgato la Legge sul progetto Bioshield del 2004. La legge prevedeva stanziamenti di miliardi di dollari per l’acquisto di vaccini in preparazione a un attacco bioterroristico e per l’accumulo di contromisure di emergenza. Per poter agire rapidamente in caso di emergenza, il Congresso ha consentito alla FDA di autorizzare prodotti formalmente non approvati per l'uso di emergenza contro una minaccia alla salute e alla sicurezza pubblica (soggetto a una dichiarazione di emergenza da parte dell'HHS). IL record indica che il Congresso si è concentrato specificamente sulla minaccia del bioterrorismo, non sulla preparazione per una pandemia naturale.
I formulazione della legge EUA sottolinea il fatto che era destinato all'uso in situazioni che implicavano armi di distruzione di massa. Ecco le 4 situazioni in cui è possibile rilasciare l'EUA:
- una determinazione da parte del Segretario per la Sicurezza Nazionale che esiste un'emergenza interna, o un potenziale significativo per un'emergenza interna, che comporta un rischio elevato di attacco con uno o più agenti biologici, chimici, radiologici o nucleari;
- una determinazione da parte del Segretario della Difesa che esiste un'emergenza militare, o un potenziale significativo per un'emergenza militare, che comporta un rischio maggiore per gli Stati Uniti stati forze militari, compreso il personale che opera sotto l'autorità del Titolo 10 o del Titolo 50, di attacco con:
- uno o più agenti biologici, chimici, radiologici o nucleari; O
- uno o più agenti che potrebbero causare, o essere altrimenti associati a, un rischio specifico e di imminente pericolo di vita per United stati forze militari;
- una determinazione da parte del segretario che esiste un'emergenza sanitaria pubblica, o un potenziale significativo per un'emergenza sanitaria pubblica, che colpisce, o ha un potenziale significativo di influenzare, la sicurezza nazionale o la salute e la sicurezza degli Stati Uniti stati cittadini che vivono all'estero e che coinvolgono uno o più agenti biologici, chimici, radiologici o nucleari, o una malattia o condizione che può essere attribuibile a tale agente o agenti; O
- l'identificazione di una minaccia materiale ai sensi della sezione 319F–2 del Legge sul servizio sanitario pubblico [42 USC 247d-6b] sufficienti a pregiudicare la sicurezza nazionale o la salute e la sicurezza dello United stati cittadini residenti all'estero.
Da nessuna parte in queste quattro situazioni si fa menzione di un’epidemia naturale, di una pandemia o di qualsiasi altro tipo di situazione di salute pubblica che non sia causata da “agenti biologici, chimici, radiologici o nucleari”.
La SARS-CoV-2 potrebbe qualificarsi come tale agente?
Se cerchi la definizione di “agenti biologici"nel Codice legale degli Stati Uniti, seguirai il seguente percorso:
Reati e procedura penale -> Reati -> Armi biologiche -> Definizioni
Pertanto, nel contesto della legge degli Stati Uniti, il termine “agenti biologici” significa armi biologiche e l’uso di tali agenti/armi è considerato un crimine.
Wikipedia lo fornisce definizione:
Un agente biologico (chiamato anche bioagente, agente di minaccia biologica, agente di guerra biologica, arma biologica o arma biologica) è un batterio, virus, protozoi, parassita, fungo, o tossina che può essere utilizzata intenzionalmente come arma bio terrorismo or guerra biologica (BN).
Su quale base giuridica è stata rilasciata l'EUA per i vaccini anti-Covid mRNA?
Sembrerebbe, sulla base delle leggi relative all'EUA, che nessuna delle quattro possibili situazioni descritte nella legge potrebbe essere applicata a un prodotto destinato a prevenire o curare una malattia causata da un agente patogeno naturale.
Tuttavia, questa legge è stata utilizzata per autorizzare i vaccini Covid a mRNA.
Date le quattro scelte elencate nella legge EUA, quella utilizzata per le “contromisure” Covid era
C) una determinazione dell'art segretario che esiste un'emergenza sanitaria pubblica, o un potenziale significativo per un'emergenza sanitaria pubblica, che colpisce, o ha un potenziale significativo di influenzare, la sicurezza nazionale o la salute e la sicurezza degli Stati Uniti stati cittadini che vivono all'estero, e che coinvolga uno o più agenti biologici, chimici, radiologici o nucleari, o una malattia o condizione che possa essere attribuibile a tale agente o agenti.
Quando applicata specificatamente al Covid, ecco come era scritto:
il Segretario del Dipartimento della Salute e dei Servizi Umani (HHS) ha stabilito che esiste un'emergenza sanitaria pubblica che ha un potenziale significativo di incidere sulla sicurezza nazionale o sulla salute e la sicurezza dei cittadini degli Stati Uniti che vivono all'estero e che coinvolge il virus che causa il Coronavirus Malattia 2019 (COVID-19)...
Non c’è dubbio che “il virus che causa il COVID-19” è considerato l’equivalente di “uno o più agenti biologici, chimici, radiologici o nucleari”.
È anche importante notare che la “determinazione di un’emergenza sanitaria pubblica” da parte dell’EUA è completamente separata e non dipende in alcun modo da qualsiasi altra dichiarazione di emergenza sanitaria pubblica, come quelle rilasciate dall’OMS, il governo degli Stati Uniti. , e il Presidente all'inizio della pandemia di Covid-19.
Quindi, anche quando l’OMS, il governo degli Stati Uniti e il Presidente dichiarano che la pandemia è finita, può ancora esserci un’autorizzazione all’uso di emergenza se il segretario dell’HHS continua a sostenere che esiste la situazione descritta nella sezione C).
Guardando tutti gli EUA per centinaia di prodotti medici correlati al Covid, è molto difficile vedere come il segretario dell’HHS possa giustificare l’affermazione secondo cui “esiste un’emergenza sanitaria pubblica che ha un potenziale significativo di influenzare la sicurezza nazionale o la salute e la sicurezza dei cittadini statunitensi che vivono all’estero” nella maggior parte, se non in tutti, di questi casi.
Ulteriori “criteri statutari” affinché la FDA conceda l’autorizzazione all’uso di emergenza
Una volta che il segretario dell’HHS dichiara che esiste un’emergenza sanitaria pubblica che giustifica l’EUA, sulla base di una delle quattro situazioni elencate nella legge, ci sono altri quattro “criteri statutari” che devono essere soddisfatti affinché la FDA possa rilasciare l’EUA . Ecco come la FDA spiega questi requisiti:
- Malattia o condizione grave o pericolosa per la vita
Affinché la FDA possa rilasciare un EUA, gli agenti CBRN a cui si fa riferimento nella dichiarazione EUA del segretario dell'HHS devono essere in grado di causare una malattia o una condizione grave o pericolosa per la vita.
NOTA: questo criterio ripete la specificazione di un agente CBRN, che è legalmente definito come un'arma utilizzata per commettere un crimine.
- Prove di efficacia
I prodotti medici che possono essere presi in considerazione per un EUA sono quelli che "potrebbero essere efficaci" per prevenire, diagnosticare o trattare malattie o condizioni gravi o pericolose per la vita che possono essere causate da uno o più agenti CBRN identificati nella dichiarazione di conformità del segretario dell'HHS. emergenza o minaccia di emergenza ai sensi della sezione 564(b).
Lo standard “può essere efficace” per gli EUA prevede un livello di prova inferiore rispetto allo standard di “efficacia” utilizzato dalla FDA per l’approvazione dei prodotti. La FDA intende valutare caso per caso la potenziale efficacia di un possibile prodotto EUA utilizzando un'analisi rischio-beneficio, come spiegato di seguito.
[GRESETTO AGGIUNTO]
QUESTIONE LEGALE: Come si può legalmente affermare che un prodotto autorizzato dall'EUA è "sicuro ed efficace" se lo standard legale per l'EUA è "può essere efficace" e la FDA dichiara che si tratta di un "livello di prova inferiore" rispetto allo standard utilizzato per le regolari approvazioni dei prodotti?
- Analisi rischio-beneficio
Un prodotto può essere preso in considerazione per un EUA se il Commissario determina che i benefici noti e potenziali del prodotto, quando utilizzato per diagnosticare, prevenire o trattare la malattia o condizione identificata, superano i rischi noti e potenziali del prodotto.
Nel determinare se i benefici noti e potenziali del prodotto superano i rischi noti e potenziali, la FDA intende guardare alla totalità delle prove scientifiche per effettuare una determinazione complessiva del rapporto rischio-beneficio. Tali prove, che potrebbe sorgere da una varietà di fonti, Può includere (ma non limitato a): risultati di studi clinici nazionali ed esteri, dati di efficacia in vivo da modelli animali e dati in vitro, disponibile per l'esame della FDA. La FDA valuterà anche la qualità e la quantità del prodotto prove disponibili, considerato lo stato attuale delle conoscenze scientifiche.
[GRESETTO AGGIUNTO]
NOTA LEGALE: non esiste uno standard legale e non esistono definizioni legali su cosa significhi che i "benefici noti e potenziali" superano i "rischi noti e potenziali". Inoltre, non esiste una definizione giuridica qualitativa o quantitativa di ciò che costituisce una “evidenza disponibile” accettabile su cui “potrebbe basarsi” l’analisi rischio-beneficio. Potrebbero non esserci prove reali, ma la convinzione che un prodotto abbia molti benefici potenziali e non molti rischi potenziali, e ciò soddisferebbe questo “requisito legale”.
- Nessuna alternativa
Affinché la FDA possa rilasciare un EUA, non deve esistere un'alternativa adeguata, approvata e disponibile al prodotto candidato per diagnosticare, prevenire o trattare la malattia o la condizione. Un potenziale prodotto alternativo può essere considerato “non disponibile” se le scorte dell’alternativa approvata non sono sufficienti per soddisfare pienamente il bisogno di emergenza.
DOMANDA LEGALE: A parte l’enorme e potenzialmente criminale diffamazione/messa al bando di trattamenti alternativi al Covid-19 come l’ivermectina e l’idrossiclorochina, a che punto è stata approvata un’alternativa per “prevenire il Covid-19” (l’unica cosa per cui sono stati acquistati i vaccini a mRNA? ) – Paxlovid, ad esempio – che renderebbe non più legale una EUA per i vaccini a mRNA?
Ecco come tutti questi “criteri statutari” sono stati effettivamente soddisfatti Autorizzazione all'uso in emergenza dei vaccini BioNTEch/Pfizer Covid mRNA:
Ho concluso che l'uso di emergenza del vaccino Pfizer-BioNTech COVID‑19 per la prevenzione del COVID-19 quando somministrato come descritto nell'ambito dell'autorizzazione (sezione II) soddisfa i criteri per il rilascio di un'autorizzazione ai sensi della sezione 564(c) del la legge, perché:
- SARS-CoV-2 può causare una malattia o una condizione grave o pericolosa per la vita, inclusa una grave malattia respiratoria, negli esseri umani infettati da questo virus;
- Sulla base della totalità delle prove scientifiche a disposizione della FDA, è ragionevole ritenere che il vaccino Pfizer-BioNTech COVID‑19 potrebbe essere efficace nella prevenzione del COVID-19e che, se utilizzato alle condizioni descritte nella presente autorizzazione, i benefici noti e potenziali del vaccino Pfizer-BioNTech COVID‑19 quando utilizzato per prevenire il COVID-19 superano i rischi noti e potenziali; E
- Non esiste un’alternativa adeguata, approvata e disponibile all’uso di emergenza del vaccino Pfizer-BioNTech COVID‑19 per prevenire il COVID-19.
[GRESETTO AGGIUNTO]
NOTA: L’unico contesto in cui la FDA ha valutato i potenziali benefici e rischi del vaccino e in cui ha stabilito che “potrebbe essere efficace” è stato nella prevenzione del Covid-19.
Non vi è alcuna considerazione, nessuna prova di benefici effettivi o potenziali, e nessuna determinazione che esista una potenziale efficacia affinché il vaccino possa fare qualcos’altro, tra cui: ridurre il rischio di malattie gravi, ridurre il rischio di ospedalizzazione, ridurre il rischio di morte , riducendo il rischio di qualsiasi condizione effettivamente o potenzialmente correlata al Covid-19.
PERTANTO, si potrebbe ragionevolmente mettere in dubbio la legalità di qualsiasi affermazione secondo cui il vaccino è “sicuro ed efficace” in un contesto diverso da “quando usato per prevenire il COVID-19” – cosa che si sapeva che i vaccini NON FANNO subito dopo la loro somministrazione. introdotto.
Se alle persone venisse detto che i vaccini a mRNA BioNTech/Pfizer sono “sicuri ed efficaci” in qualsiasi cosa diversa dalla prevenzione del Covid-19, e se fossero minacciate di conseguenze per la mancata assunzione del vaccino per qualsiasi cosa diversa dalla prevenzione del Covid-19, potrebbero hanno una argomentazione legittima secondo cui sono stati illegalmente costretti a prendere un prodotto non approvato in base a dichiarazioni fraudolente?
Requisiti di terzo livello per EUA per prodotti non approvati
Una volta ottenuta la dichiarazione di emergenza specifica per l’EUA e una volta che la FDA dichiara che il prodotto può essere efficace e che qualsiasi prova disponibile (da zero a infinito) dimostra che i suoi benefici superano i rischi (come determinato da ciò che la FDA pensa che potrebbero be), esiste un ulteriore livello di regolamentazione non correlata alla sicurezza e all’efficacia.
Ecco come a Rapporto 2018 del Servizio di ricerca del Congresso sull'EUA spiega questo:
L'FFDCA §564 impone alla FDA di imporre determinate condizioni richieste in un EUA e consente ulteriori condizioni discrezionali ove appropriato. Le condizioni richieste variano a seconda che l'EUA riguardi un prodotto non approvato o un uso non approvato di un prodotto approvato. Per un prodotto non approvato, le condizioni d'uso devono:
(1) garantire che gli operatori sanitari che somministrano il prodotto ricevano le informazioni richieste;
(2) garantire che le persone a cui viene somministrato il prodotto ricevano le informazioni richieste;
(3) prevedere il monitoraggio e la segnalazione degli eventi avversi associati al prodotto; E
(4) prevedere la tenuta dei registri e la rendicontazione da parte del produttore.
QUESTIONE LEGALE: Quali sono esattamente le “informazioni richieste?” Sappiamo che le persone sono state informate che i vaccini avevano ricevuto l’autorizzazione all’uso di emergenza. Ma è stato detto loro che ciò significa “un livello di prova inferiore” rispetto a quello richiesto per affermazioni “sicure ed efficaci” su altri prodotti medici? Sono stati informati che esistono diversi livelli di “sicuro ed efficace” a seconda che un prodotto abbia un EUA o un altro tipo di autorizzazione?
NOTA: la legge richiede che esista un modo per monitorare e segnalare eventi avversi. Tuttavia, non indica chi effettua il monitoraggio, quali sono gli standard per la rendicontazione e qual è la soglia per intraprendere azioni sulla base delle segnalazioni.
EUA rispetto a ogni altro percorso di approvazione di farmaci/vaccini
Come ricercatore/scrittore Sasha Latypova ha sottolineato, molte persone erano confuse da EUA, perché assomiglia molto a EAU, che sta per "Expanded Access Use". Si tratta di un tipo di autorizzazione concessa ai prodotti medici quando ce n'è urgente bisogno da parte di un particolare gruppo di pazienti (ad esempio, pazienti affetti da cancro allo stadio IV la cui aspettativa di vita è misurata in mesi) che sono disposti a rischiare eventi avversi e persino la morte in cambio dell'accesso. ad un trattamento sperimentale.
L'Autorizzazione all'Uso di Emergenza non è in alcun modo correlata, né ha alcuna somiglianza con, l'Utilizzo dell'Accesso Esteso.
I vari percorsi legali per l'autorizzazione dei prodotti medici sono presentati in modo chiaro in una tabella evidenziata dal ricercatore legale Caterina Watt. La tabella fa parte di una presentazione del 2020 per una sessione di apprendimento congiunto FDA-CDC: Aggiornamenti normativi sull'uso delle contromisure mediche.
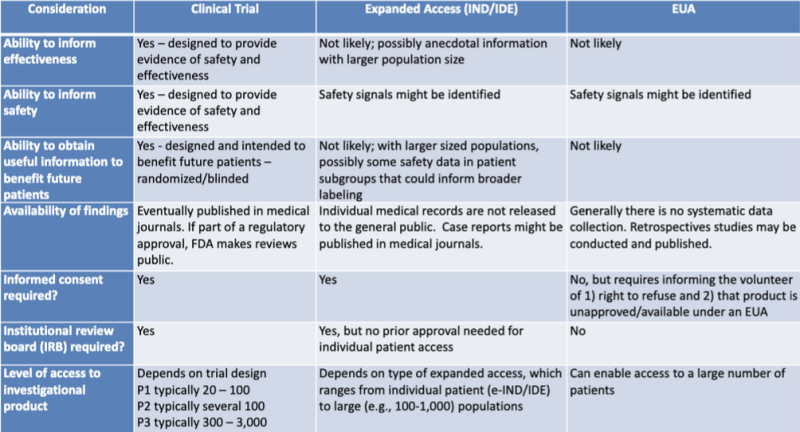
Questa tabella mostra molto chiaramente che è improbabile che il processo EUA fornisca informazioni sull'efficacia del prodotto, non è progettato per fornire prove di sicurezza, non è probabile che fornisca informazioni utili a beneficio dei futuri pazienti, non comporta una raccolta sistematica di dati, non richiede studi retrospettivi, nessun consenso informato e nessun comitato di revisione istituzionale.
Inoltre, in a 2009 Pubblicazione accademica nazionale dell'Istituto di Medicina, evidenziato anche da Watt, intitolato “Medical Countermeasures: Dispensing Emergency Use Authorization and the Postal Model – Workshop Summary” troviamo tale affermazione a p. 28:
È importante riconoscere che una EUA non fa parte del percorso di sviluppo; è un'entità completamente separata che viene utilizzata solo durante le situazioni di emergenza e non fa parte del processo di approvazione del farmaco.
Ciò significa che le approvazioni delle contromisure Covid-19 basate sugli EUA erano illegali? Significa che non esiste un modo legale per affermare che un prodotto EUA sia “sicuro ed efficace” perché NON FA PARTE DEL PROCESSO DI APPROVAZIONE DEL FARMACO?
Conclusione
Ciò è del tutto evidente, date tutte le informazioni contenute in questo articolo e in quello precedente Parte 1, che i vaccini BioNTach/Pfizer Covid mRNA sono stati sviluppati, prodotti e autorizzati in base alle leggi militari riservate a situazioni di emergenza che coinvolgono guerra biologica/terrorismo, e non a malattie naturali che colpiscono l’intera popolazione civile.
Pertanto, il rispetto delle normative e della supervisione che ci aspettiamo di trovare quando un prodotto è ritenuto “sicuro ed efficace” per l’intera popolazione civile non era richiesto dalla legge.
Questa analisi può essere utilizzata per contestare la legalità della dichiarazione “sicura ed efficace” da parte di quei funzionari governativi che sapevano cosa comportava l’EUA? Ci sono altre implicazioni legali?
Lo spero.
È importante sottolineare che nelle contestazioni legali contro i vaccini Covid mRNA portate finora, non ci sono state sentenze (di cui sono a conoscenza) sulla possibilità o meno di applicare il diritto militare, come l’OTA e l’EUA, a situazioni civili. Tuttavia, c'è stata una dichiarazione del giudice della Corte distrettuale Michael Truncale, nella sua archiviazione del caso del segnalante Brook Jackson contro Ventavia e Pfizer, è importante tenerlo presente.
Qui il giudice riconosce che l’accordo per i vaccini a mRNA BioNTech/Pfizer era un OTA militare, ma si rifiuta di pronunciarsi sulla sua applicabilità a circostanze non militari (malattie naturali, 100 milioni di dosi per lo più non per uso militare) in base alle quali è stato rilasciato:
Il fatto che sia il personale militare che i civili abbiano ricevuto il vaccino non indica che l’acquisizione del vaccino fosse irrilevante per migliorare l’efficacia della missione militare. Ancora più importante, la signora Jackson sta in effetti chiedendo a questa Corte di annullare la decisione del Dipartimento della Difesa di esercitare l’Other Transaction Authority per acquistare il vaccino della Pfizer. Ma come la Corte Suprema degli Stati Uniti ha da tempo sottolineato, le “decisioni complesse, sottili e professionali riguardanti la composizione, l’addestramento, l’equipaggiamento e il controllo di una forza militare sono essenzialmente giudizi militari professionali”. Gilligan contro Morgan, 413 USA 1, 10 (1973). Pertanto, è “difficile concepire un’area di attività governativa in cui i tribunali abbiano meno competenza”. Id. Questa Corte non porrà il veto alle sentenze del Dipartimento della Difesa riguardanti l’efficacia della missione durante un’emergenza nazionale.
Questo è solo uno dei tanti ostacoli legali che rimangono nella battaglia per mettere definitivamente al bando tutti i prodotti mRNA approvati durante l’emergenza Covid-19 e tutti i successivi prodotti mRNA la cui approvazione era basata sul processo di approvazione Covid-19.
Pubblicato sotto a Licenza internazionale Creative Commons Attribution 4.0
Per le ristampe, reimpostare il collegamento canonico all'originale Istituto di arenaria Articolo e Autore.








